Descriptions
TOFAJAK (Tofacitinib) is formulated with the citrate salt of tofacitinib, a JAK inhibitor.
Tofacitinib citrate is a white to off-white powder with the following chemical name:
( 3 R , 4 R ) – 4 – m e t h y l – 3 – ( m e t h y l – 7 H – p y r r o l o [2,3-d]pyrimidin-4-ylamino)-ß-oxo-1piperidinepropanenitrile, 2-hydroxy-1,2,3-propanetricarboxylate (1:1).
The solubility of tofacitinib citrate in water is 2.9 mg/mL. Tofacitinib citrate has a molecular weight of 504.5 Daltons (or 312.4 Daltons as the tofacitinib free base) and a molecular formula of C16H20N6O•C6H8O7. The chemical structure of tofacitinib citrate is:

CLINICAL PHARMACOLOGY
Mechanism of Action:
Tofacitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Tofacitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. JAK enzymes transmit cytokine signaling through pairing of JAKs (e.g., JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibited the in vitro activities of JAK1/JAK2, JAK1/JAK3, and JAK2/JAK2 combinations with IC50 of 406, 56, and 1377 nM, respectively. However, the relevance of specific JAK combinations to therapeutic effectiveness is not known.
Treatment with TOFACITINIB was associated with dose-dependent reductions of circulating CD16/56+ natural killer cells, with estimated maximum reductions occurring at approximately 8-10 weeks after initiation of therapy. These changes generally resolved within 2-6 weeks after discontinuation of treatment. Treatment with TOFACITINIB was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent. The clinical significance of these changes is unknown. Total serum IgG, IgM, and IgA levels after 6-month dosing in patients with rheumatoid arthritis were lower than placebo; however, changes were small and not dose-dependent. After treatment with TOFACITINIB in patients with rheumatoid arthritis, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with TOFACITINIB treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the pharmacokinetic half-life. Similar changes in T cells, B cells, and serum CRP have been observed in patients with active psoriatic arthritis although reversibility was not assessed. Total serum immunoglobulins were not assessed in patients with active psoriatic arthritis.
Pharmacodynamics:
Pharmacokinetics:
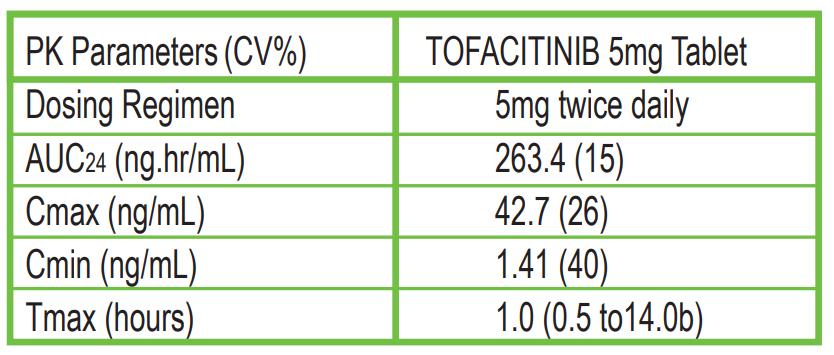
Following oral administration of TOFACITINIB, peak plasma concentrations are reached within 0.5-1 hour, elimination half-life is about 3 hours and a dose-proportional increase in systemic exposure was observed in the therapeutic dose range. Steady state concentrations are achieved in 24-48 hours with negligible accumulation after twice daily administration.
Table: Pharmacokinetic Parameters of TOFACITINIB Following Oral Dosing
Absorption
The absolute oral bioavailability of TOFACITINIB is 74%. Coadministration of TOFACITINIB with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, TOFACITINIB was administered without regard to meals.
After intravenous administration, the volume of distribution is 87 L. The protein binding of tofacitinib is approximately 40%. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma. Metabolism and Excretion Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule.
Distribution
INDICATION AND USAGE:
TOFAJAK is a Janus kinase (JAK) inhibitor indicated for :
- Rheumatoid Arthritis: TOFAJAK is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate.
- Limitations of Use: Use of TOFAJAK in combination with biologic DMARDs or potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
- Psoriatic Arthritis: TOFAJAK is indicated for the treatment of adult patients with active psoriatic arthritis who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs).
- Limitations of Use: Use of TOFAJAK in combination with biologic DMARDs or potent immunosuppressants such as azathioprine and cyclosporine is not recommended. (1)
- Ulcerative Colitis: TOFAJAK is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis (UC), who have had an inadequate response or who are intolerant to TNF blockers.
- Limitations of Use: Use of TOFAJAK in combination with biological therapies for UC or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
CONTRAINDICATIONS:
None

DOSAGE ADMINISTRATION:
Recommended Dosage
Rheumatoid Arthritis
- TOFAJAK 5 mg twice daily.
- Recommended dosage in patients with moderate and severe renal impairment or moderate hepatic impairment is TOFAJAK 5 mg once daily.
Psoriatic Arthritis (in combination with nonbiologic DMARDs)
- TOFAJAK 5 mg twice daily.
- Recommended dosage in patients with moderate and severe renal impairment or moderate hepatic impairment is TOFAJAK 5 mg once daily.
- Induction: 2 tablets of TOFAJAK 5 mg twice daily for 8 weeks; evaluate patients and transition to maintenance therapy depending on therapeutic response. If needed, continue 2 tablets of TOFAJAK 5 mg twice daily for a maximum of 16 weeks. Discontinue after 16 weeks if adequate therapeutic response is not achieved.
- Maintenance: TOFAJAK 5 mg twice daily. For patients with loss of response during maintenance treatment, 2 tablets of TOFAJAK 5 mg twice daily may be considered and limited to the shortest duration, with careful consideration of the benefits and risks for the individual patient. Use the lowest effective dose needed to maintain response.
- Dosage adjustment is needed in patients with moderate and severe renal impairment or moderate hepatic impairment.
Ulcerative Colitis
WARNINGS AND PRECAUTIONS
SERIOUS INFECTIONS
Patients treated with TOFAJAK are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt TOFAJAK until the infection is controlled.
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before TOFAJAK use and during therapy. Treatment for latent infection should be initiated prior to TOFAJAK use.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized,disease.
- Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
Reported infections include:
MORTALITY
Rheumatoid arthritis patients 50 years of age and older with at least one cardiovascular (CV) risk factor treated with TOFACITINIB 10 mg twice a day had a higher rate of all-cause mortality, including sudden CV death, compared to those treated with TOFACITINIB 5 mg given twice daily or TNF blockers in a large, ongoing, postmarketing safety study. MALIGNANCIES Lymphoma and other malignancies have been observed in patients treated with TOFACITINIB. Epstein Barr Virus-associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with TOFACITINIB and concomitant immunosuppressive medications.
Thrombosis, including pulmonary embolism, deep venous thrombosis, and arterial thrombosis have occurred in patients treated with TOFAJAK and other Janus kinase inhibitors used to treat inflammatory conditions. Rheumatoid arthritis patients who were 50 years of age and older with at least one CV risk factor treated with TOFACITINIB 10 mg twice daily compared to TOFACITINIB 5 mg twice daily or TNF blockers in a large, ongoing postmarketing safety study had an observed increase in incidence of these events. Many of these events were serious and some resulted in death. Avoid TOFACITINIB in patients at risk. Discontinue TOFACITINIB and promptly evaluate patients with symptoms of thrombosis. For patients with ulcerative colitis, use TOFACITINIB at the lowest effective dose and for the shortest duration needed to achieve/maintain therapeutic response.
THROMBOSIS
ADVERSE REACTIONS
Most common adverse reactions are:
- Rheumatoid and Psoriatic Arthritis: Reported during the first 3 months in rheumatoid arthritis controlled clinical trials and occurring in ≥2% of patients treated with TOFACITINIB monotherapy or in combination with DMARDs: upper respiratory tract infection, nasopharyngitis, diarrhea, and headache.
- Ulcerative Colitis: Reported in ≥5% of patients treated with either 5 mg or 10 mg twice daily of TOFACITINIB and ≥1% greater than reported in patients receiving placebo in either the induction or maintenance clinical trials nasopharyngitis, elevated cholesterol levels, headache, upper respiratory tract infection, increased blood creatine phosphokinase, rash, diarrhea, and herpes zoster.
- Polyarticular Course Juvenile Idiopathic Arthritis: Consistent with common adverse reactions reported in adult rheumatoid arthritis patients.
Strong CP3A4 Inhibitors (e.g., ketoconazole) Increased exposure to tofacitinib; Dosage adjustment is recommended
Moderate CYP3A4 Inhibitors Coadministered with Strong CYP2C19 Inhibitors (e.g., fluconazole)
Increased exposure to tofacitinib; Dosage adjustment is recommended
Strong CYP3A4 Inducers (e.g., rifampin)
Decreased exposure to tofacitinib and may result in loss of or reduced clinical response; Coadministration is not recommended
Immunosuppressive Drugs (e.g., azathioprine, tacrolimus, cyclosporine)
Risk of added immunosuppression; coadministration with biologic DMARDs or potent immunosuppressants has not been studied in patients with rheumatoid arthritis, psoriatic arthritis, UC, or pcJIA.; Coadministration is not recommended.
DRUG INTERACTIONS:
USE IN SPECIFIC POPULATIONS
Pregnancy
Available data with TOFACITINIB use in pregnant women are insufficient to establish a drug associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the mother and the fetus associated with rheumatoid arthritis and UC in pregnancy. Disease-Associated Maternal and/or Embryo/Fetal Risk Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
There are no data on the presence of tofacitinib in human milk, the effects on a breastfed infant, or the effects on milk production. Tofacitinib is present in the milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Given the serious adverse reactions seen in patients treated with TOFACITINIB, such as increased risk of serious infections, advise patients that breastfeeding is not recommended during treatment and for at least 18 hours after the last dose of TOFACITINIB.
Lactation
Females and Males of Reproductive Potential Contraception Females
In an animal reproduction study, tofacitinib at AUC multiples of 13 times the recommended dose of 5 mg twice daily and 6.3 times the maximum recommended dose of 10 mg twice daily demonstrated adverse embryo-fetal findings. However, there is uncertainty as to how these animal findings relate to females of reproductive potential treated with the recommended clinical dose. Consider pregnancy planning and prevention for females of reproductive potential. Infertility Females Based on findings in rats, treatment with TOFACITINIB may result in reduced fertility in females of reproductive potential. It is not known if this effect is reversible.
Of the 3315 patients who enrolled in rheumatoid arthritis Studies I to V, a total of 505 rheumatoid arthritis patients were 65 years of age and older, including 71 patients 75 years and older. The frequency of serious infection among TOFACITINIB-treated subjects 65 years of age and older was higher than among those under the age of 65. Of the 1156 TOFACITINIB-treated patients in the UC program, a total of 77 patients (7%) were 65 years of age or older. The number of patients aged 65 years and older was not sufficient to determine whether they responded differently from younger patients.
Geriatric Use:
Renal Impairment
Moderate and Severe Impairment TOFACITINIB-treated patients with moderate or severe renal impairment had greater tofacitinib blood concentrations than TOFACITINIB-treated patients with normal renal function. Therefore, dosage adjustment of TOFACITINIB is recommended in patients with moderate or severe renal impairment (including but not limited to those with severe insufficiency who are undergoing hemodialysis) Mild impairment: No dosage adjustment is required in patients with mild renal impairment.
Severe Impairment: TOFACITINIB has not been studied in patients with severe hepatic impairment; therefore, use of TOFACITINIB in patients with severe hepatic impairment is not recommended. Moderate Impairment: TOFACITINIB-treated patients with moderate hepatic impairment had greater tofacitinib blood concentration than TOFACITINIB-treated patients with normal hepatic function. Higher blood concentrations may increase the risk of some adverse reactions. Therefore, dosage adjustment of TOFACITINIB is recommended in patients with moderate hepatic impairment. Mild Impairment: No dosage adjustment of TOFACITINIB is required in patients with mild hepatic impairment. Hepatitis B or C Serology The safety and efficacy of TOFACITINIB have not been studied in patients with positive hepatitis B virus or hepatitis C virus serology.
Hepatic Impairment
OVERDOSAGE:
PRESENTATION:
INSTRUCTIONS:
There is no specific antidote for overdose with TOFACITINIB. In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions.
TOFAJAK 5mg tablets are available in bottle pack of 10’s.
Store below 30°C in a dry place, protect from light. Keep out of the reach of children. To be sold on the prescription of a registered medical practitioner only. Use as directed by the Physician.